
TGA分类及认证流程
作者: 浏览次数:23412 日期:2023-08-29
什么是澳大利亚TGA认证?
根据澳大利亚医疗用品法(Therapeutic Goods Act 1989)规定,所有在澳大利亚上市的医疗用品(药品和医疗器械)必须按有关要求,向澳大利亚医疗用品管理局(Therapeutic Goods Administration, TGA)提出注册或登记申请,获得注册登记(Australian Register of Therapeutic Goods,ARTG)后才能合法上市。
01.澳大利亚TGA认证的好处
直接获得发达国家澳大利亚的GMP认证证书;
直接获得与澳大利亚有GMP互认(MRA)的26个国家的GMP认可;
药品企业可以获得国家有关“获得发达国家注册认证产品优惠政策”;
注册认证过程将实质性大幅提高企业产品注册与GMP管理水平;
注册认证过程将改变和提高企业的国际化理念和认识;
大幅提高企业和产品形象及美誉度,极大有利于国内市场营销;
保健食品华丽转身为发达国家批准的“药品”,极大有利于国内、国际市场营销;
易获得与澳大利亚有GMP符合确认(CV)关系的52个国家/组织的GMP认可;
易进入与澳大利亚有互认关系及PIC/s成员国的注册和市场准入;
易进入其它英联邦国家的注册和市场准入;
东南亚、非洲和拉丁美洲许多国家对澳大利亚上市许可有很高认可度。
02.主管机构及法规
- 监督机构 :TGA 是Therapeutic Goods Administration的简写,全称是医疗用品管理局,它是澳大利亚的医疗用品(包括药物、医疗器械、基因科技和血液制品)的监督机构。
- 法规要求:✔ Therapeutic Goods Act 1989-the Act;
✔ Therapeutic Goods(Medical Devices)Regulations 2002-the Regulations。
- 监管途径:上市前通知/注册。
• 授权代表:Austrialian Sponsor 澳大利亚代理(澳代)--持证人,只负责法规注册, 不涉及商品后续市场的销售,有别于经销商的角色。 - QMS 要求:Medical Device Single Audit Program (简称MDSAP)医疗器械单一审核程序。
03.产品分类
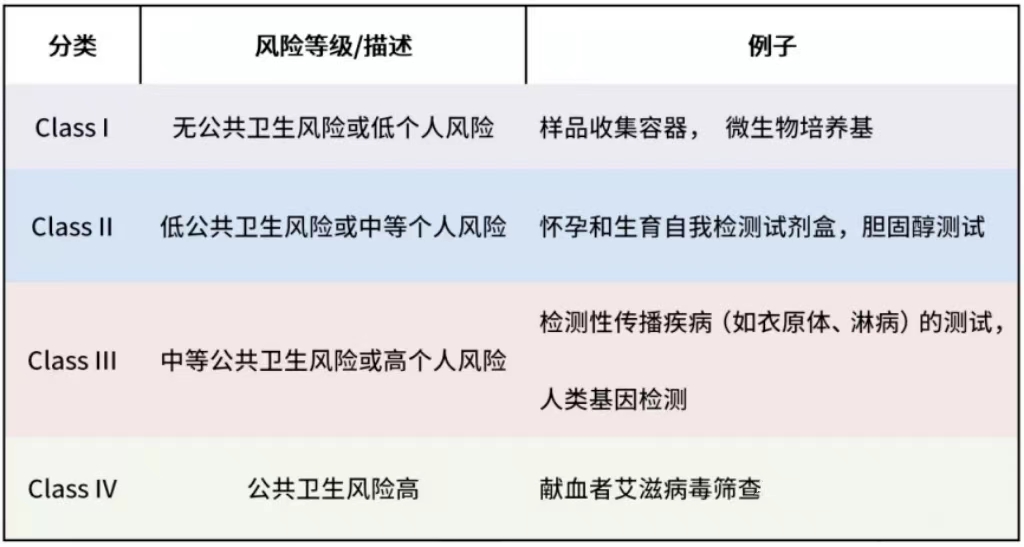
在澳洲,体外诊断产品(IVD)视为医疗器械产品(MD)的子集受到监管, 其预期目的和风险管理方法是医疗器械分类的基础,制造商负责对其医疗器械进行分类。设备的分类级别越高,设备应用的合格评定程序的要求就越高。若特定医疗器械适用多种分类规则,该设备则按照适用的最高风险等级进行分类。
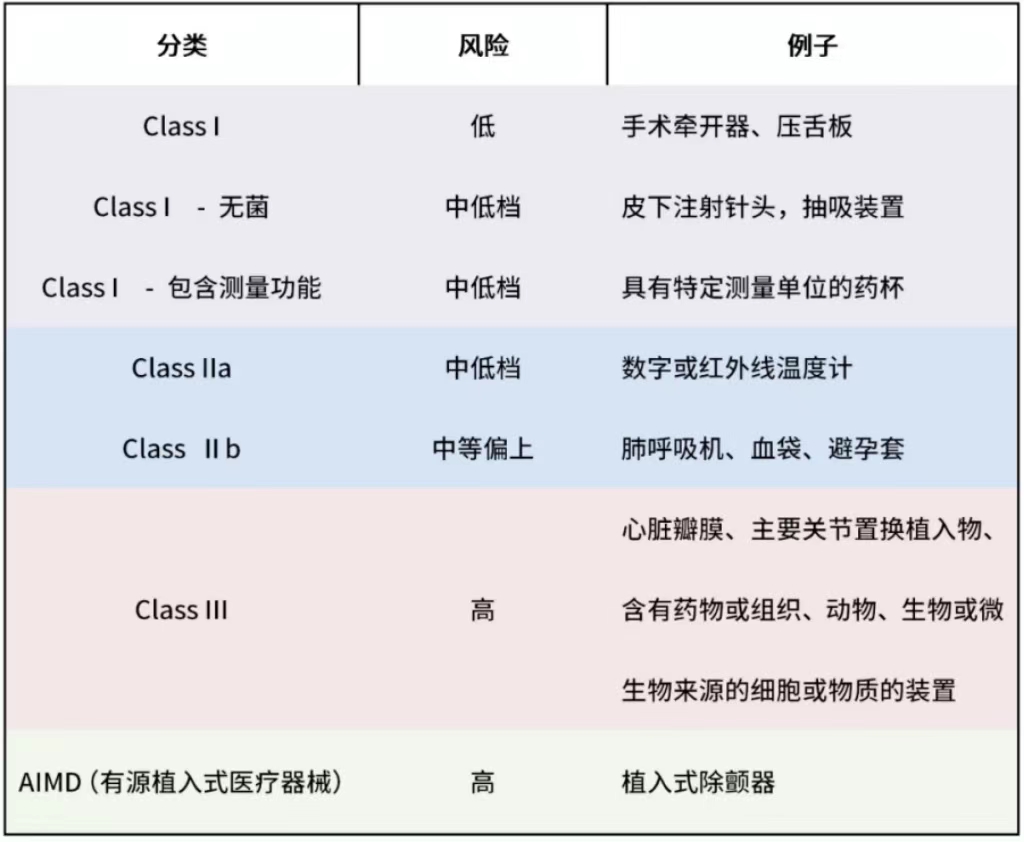
医疗器械分类(非IVD医疗器械)
除体外诊断医疗器械外,医疗器械根据其预期用途进行分类。特别是,分类规则考虑了对人体的侵入程度、使用的持续时间和位置,以及设备是否依赖于身体或重力以外的能量来源。
IVD医疗器械的分类
04.澳大利亚医疗器械注册流程
制造商和澳洲代表的医疗器械制造商的责任
制造商必须:
a.为每个医疗器械确定:
- 分类
- 预期目的
- 适当GMDN码
b.选择并应用适当的合格评定程序,以证明遵守基本原则;
c.在申请的TGA或欧盟机构的评估证据之前确保有现场的适当的程序,包括文件, 以证明遵守基本原则;
d.获得合格评定证据,并确保证书上的信息保持最新和有效;
e.支付获取证据的合格评定程序申请费和评估费;
f 准备一份澳大利亚合格声明,包括医疗器械制造的所有细节;
g.一旦获得必要的合格评定的证据后,确保其合格评定程序适当地维持,而且现行 的要求得到满足(例如,报告的不良事件,定期进行质量体系审核);
h.通知TGA器械的设计,生产或预期性能的大变更;
i .预期目的 制造商必须确定医疗器械的预期目的。预期目的是注册程序中至关重要的,在分类过程中要考虑的。
澳洲代表负责医疗器械在ARTG登记
澳洲代表在收到制造商以下信息后在ARTG登记器械:
a.器械分类
b.器械预定目的
c.GMDN代码和术语
d.合格评定认证
e.澳大利亚一致性声明
Sponsor澳州代表在澳洲以外制造商的产品进入到澳洲市场的时候扮演重要的角色。一方面作为制造商和澳洲TGA沟通的桥梁和纽带,另一方面Sponsor的信息需要体现在产品的包装上。依据法规,Sponsor只能是澳洲本土的公司,同时具备相关的法律和技术法规知识的人员才可以担任。
依据Australian Therapeutic Goods (Medical Devices) Regulations 2002,澳大利亚对医疗器械分为I类,Is and Im, IIa, IIb, III类,产品的分类几乎和欧盟分类一致,如果贵公司产品已经获得CE标志,则产品类别可以按照CE分类。如果已经获得欧盟公告机构(Notified Body)签发的CE证书,是可以被TGA认可的,并可以作为满足澳大利亚安全法规的重要注册资料。
在澳大利亚供应医疗器械程序 - 所有的I类非无菌和非测量器械
- 制造商准备必要的技术文件和澳大利亚的符合性声明
b. 主办者通过 TGA 递交在ARTG登记申请
c. 医疗器械制造商在ARTG和TGA登记后通知主办者
d. 主办者从电子业务系统打印出登记证书
e. 器械上市后持续监控
在澳大利亚供应医疗器械程序 - 如果医疗器械是在澳大利亚制造的(即I类消毒; I类测量; IIa类; IIb类,第III类,AIMD类)
- 制造商决定质量规程,用于证明器械符合相关的基本原则,并准备必要的文 件
b. 制造商申请TGA合格评定证shu
c. 制造商准备澳大利亚符合性声明
d. 主办者向TGA提交制造商的证据
e. 主办者递交在ARTG登记申请
f. 在ARTG登记后主办者可以在澳大利亚供应器械
g. 器械上市后持续监控
在澳大利亚供应医疗器械程序 - 如果医疗器械是在海外制造的(即I类消毒; I类测量; IIa类; IIb类,III类,AIMD类)
- 制造商从TGA或欧盟的认证机构获得合格评定证据
b. 制造商准备澳大利亚符合性声明
c. 主办者向TGA提交制造商的证据
d. 主办者递交在ARTG登记申请
e. 在ARTG登记后主办者可以在澳大利亚供应器械
f. 器械上市后持续监控
澳洲代表负责医疗器械在ARTG登记
- 制造商从TGA或欧盟的认证机构获得合格评定证据
b. 制造商准备澳大利亚符合性声明
c. 主办者向TGA提交制造商的证据
d. 主办者递交在ARTG登记申请
e. 在ARTG登记后主办者可以在澳大利亚供应器械
f. 器械上市后持续监控
仲邈检测可以提供一体化合规性注册认证解决方案,为客户进行国内医疗器械注册及生产许可、CE-MDR、CE-IVDR、FDA、ISO13485、MDSAP等注册认证服务。仲邈检测秉承“专业,诚信,共赢”的企业理念,专业示人,诚以待人,与业界同行合作、分享,提供超出客户期望的增值服务,携手客户、同行共成长! 更多详情请致电:400-869-7268,18101860670(微信同号),18117149592(微信同号),18116429616(微信同号)
