
在MDR法规中,对公告机构有何通用要求?
作者: 浏览次数:76412 日期:2023-08-29
a.公告机构的正常运作对于确保高水平的健康和安全保护以及公民对系统的信心至关重要。因此,成员国根据详细和严格的标准对指定机构进行的指定和监测,应在欧盟级别实行控制。
b.公告机构对制造商的技术文件的评估,特别是其临床评估文件,应由负责公告机构的主管机构进行严格评估。此类评估是一种基于风险的手段,用于监督和监测公告机构活动,评估时可采取相关文件抽样的方法。
c.应加强公告机构对制造商的监管,包括其进行突击飞行检查和对器械进行物理或实验室测试的权利和义务,以确保制造商在收到原始证书后持续合规。
新版MDR法规对公告机构提出更多更高的要求,所以在MDR法规生效之后,公告机构的数量也会有所减少,在法规中说公告机构的正常运作,对于确保高水平的健康和安全保护,以及公民对系统的信心至关重要,因此成员国根据详细和严格的标准对指定机构进行的指定和监测应在欧盟级别实行控制
第一步考虑在官网的公告机构
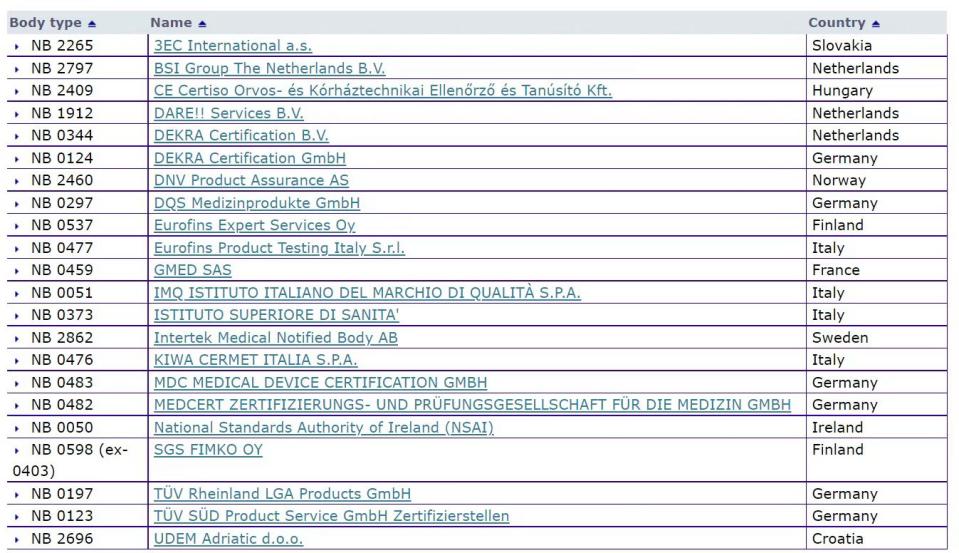
第二步考虑的如外企的总部或者是以后预期的持证方的位置也是筛选公告机构的一个考虑点
第三步就进一步缩小然后企业的清单一些具有较广审核范围覆盖所有可能产品族的公告机构是最安全的选择。然而通常呢像这样的机构也非常忙,所以审核范围较小的公告机构接受新客户就更加灵活。另外自己企业的产品组合可能需要多个公告机构支持,理解产品组合以及充分的准备就非常重要。
在今年五月份,MDR的法规正式实施了,有些公告机构会突然离开,这就意味着有些制造商可能突然失去公告机构来备案他们的器械产品,也被称为孤儿制造商。
欧盟监管当局短期内会接管证书或者直到证书失效。但是监管当局会经常向这些制造商提出一些额外的要求,以至于企业在艰难的寻找另一个公告机构时,还要应对来自监管机构的问询。
切换公告机构最快的方式就是保证企业的流程和文档的状态是非常好的,也就意味着企业需要去解决目前情况和检查所有的技术文件和临床报告,更新所有的文档甚至是非常细小的。检查上市后监管和上市后临床跟踪,制定敏捷的过渡期计划,并且根据企业产品的优先级来制定不同的项目顺序。
如果发现公告机构将结束业务,需要立刻制定紧急计划。首先计算出企业的产品还有多久不能在欧盟上市,上市的产品有两个含义,第一个是产品所有权已转移至其他法律主体,不再为企业所有。第二个是产品在欧盟境内境外制造商上市前,必须将产品运输至境内,并且有变更后产品的所有权。产品在欧盟市场上市便可以进一步合法配送。企业就会借此向欧盟的供应商备货,这样便可以帮助企业平稳的度过换证的时间间隙。
仲邈检测可以提供一体化合规性注册认证解决方案,为客户进行国内医疗器械注册及生产许可、CE-MDR、CE-IVDR、FDA、ISO13485、MDSAP等注册认证服务。仲邈检测秉承“专业,诚信,共赢”的企业理念,专业示人,诚以待人,与业界同行合作、分享,提供超出客户期望的增值服务,携手客户、同行共成长! 更多详情请致电:400-869-7268,18101860670(微信同号),18117149592(微信同号),18116429616(微信同号)
