

联系我们
仲邈检测技术(上海)有限公司
电话:400-869-7268
孔老师:18101860670
李老师:18117149592
网址:www.shzmiao.com
邮箱:sales@shzmiao.cn
地址:上海市诸光路1588弄虹桥世界中心B栋一楼763号
美国FDA注册
一、 美国监管部门
1. 美国FDA

FDA负责医疗器械的部门是医疗器械和放射健康中心CDRH(Center for Devices and Radiological Health),其职责是确保美国市场上销售的医疗器械在按照指定用途应用时在合理基础上的安全和有效,同时还负责所有放射性电子产品的安全。
CDRH的器械评估办公室ODE(Office of Device Evaluation)负责审议所有上市前的申请(Premarket Approval,简称PMA)、上市前通
告(510(K)/Premarket Notification)和上市前批准(PMA)的申请,以及所有未经批准、但将用于临床试验的医疗器械,以确保医疗器械和放射产品的安全、有效和高质量。
2. 相关法规
1976 Food, Drug, and Cosmetic Act, Medical Device Amendment
1995 Safe Medical Device Act
3. 医疗器械和IVD产品风险等级分类
根据风险等级的不同,FDA将医疗器械分为三类(Ⅰ,Ⅱ,Ⅲ),Ⅲ类风险等级最高,I类风险等级最低。FDA对每一种医疗器械都明确规定其产品分类和管理要求,目前FDA医疗器械产品目录中共有1700多种。
任何一种医疗器械想要进入美国市场,必须首先确认申请上市产品分类和管理要求。FDA是根据专家委员会的建议来最终决定医疗器械产品详细分类的,并且在定期公布分类结果的同时,每年还会对法规代码库进行适时更新。
4. 准入必要条件
制造商需要遵守FDA的质量体系法规(QSR)确保生产过程的质量和一致性。
Class I:低风险器械,对于可豁免510(k)的Class I器械只需进行企业注册、产品列名即可进入美国市场。对于其他的Class I风险等级器械,需要根据产品特性需要递交产品技术文件获得K号。
Class II:中等风险器械,对于II类可510(k)豁免的产品,注册条件同I类器械;对于其他的Class II器械需要通过510(K)来证明其安全性和有效性,少部分II类器械需要上市前批准(PMA)。
Class III:高风险器械,这些器械需要获得上市前批准(PMA),提供详细的临床试验数据和有效性证明。
所有医疗器械企业必须在FDA进行企业注册和产品列名。
5. 注册语言
英语
二、 注册流程|周期|官费
1. 注册流程
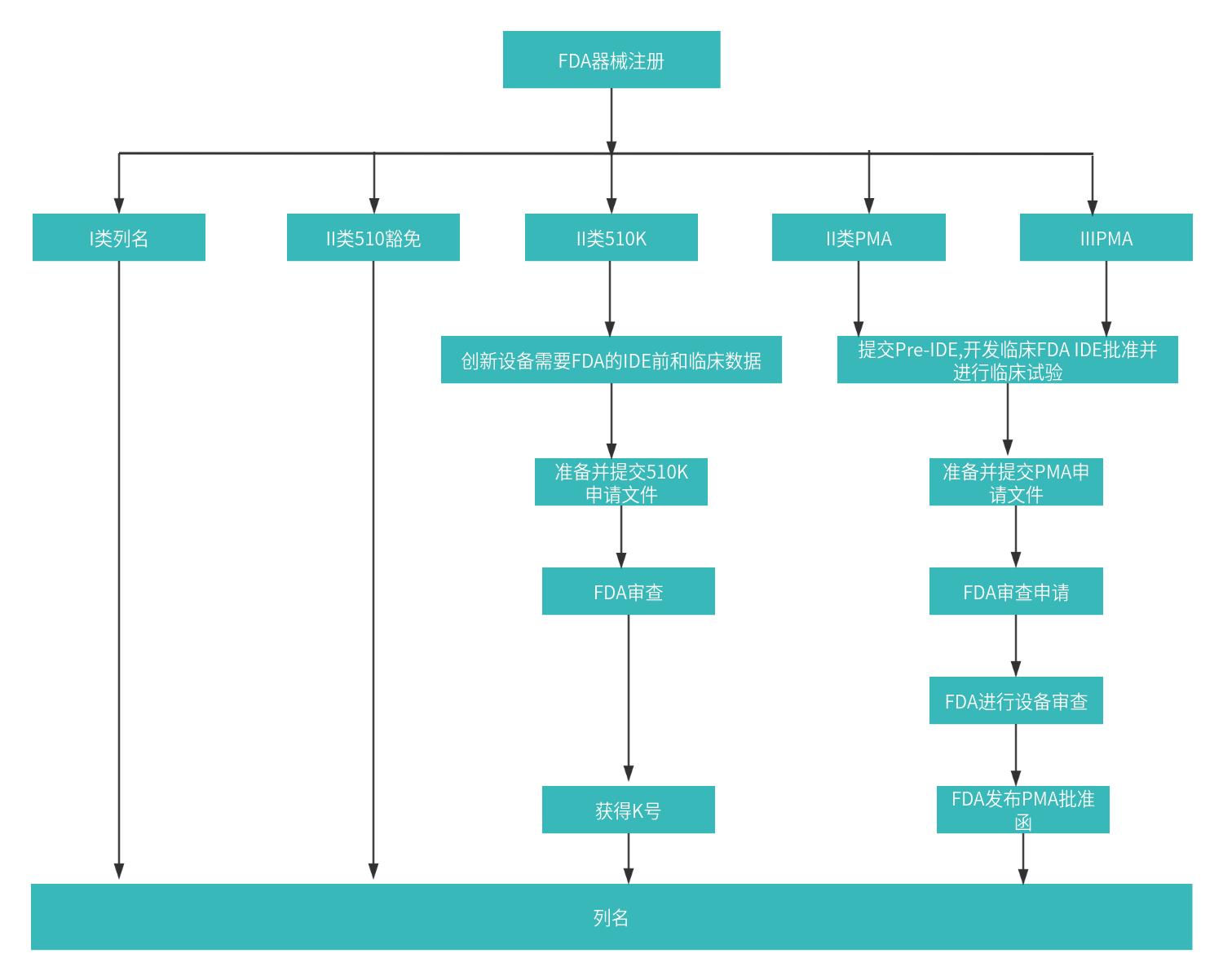
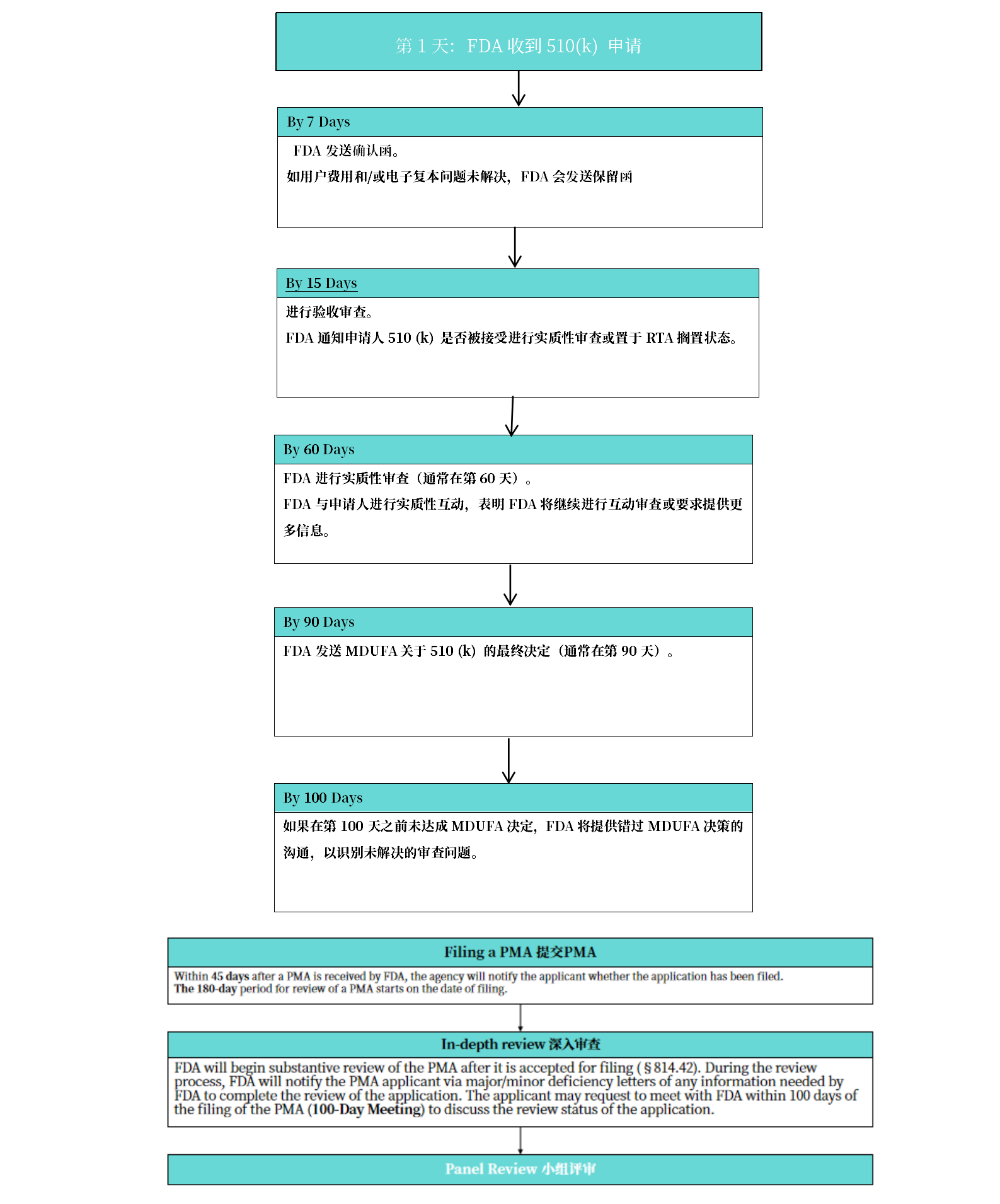
2. 注册周期
周期
3. 官方费用
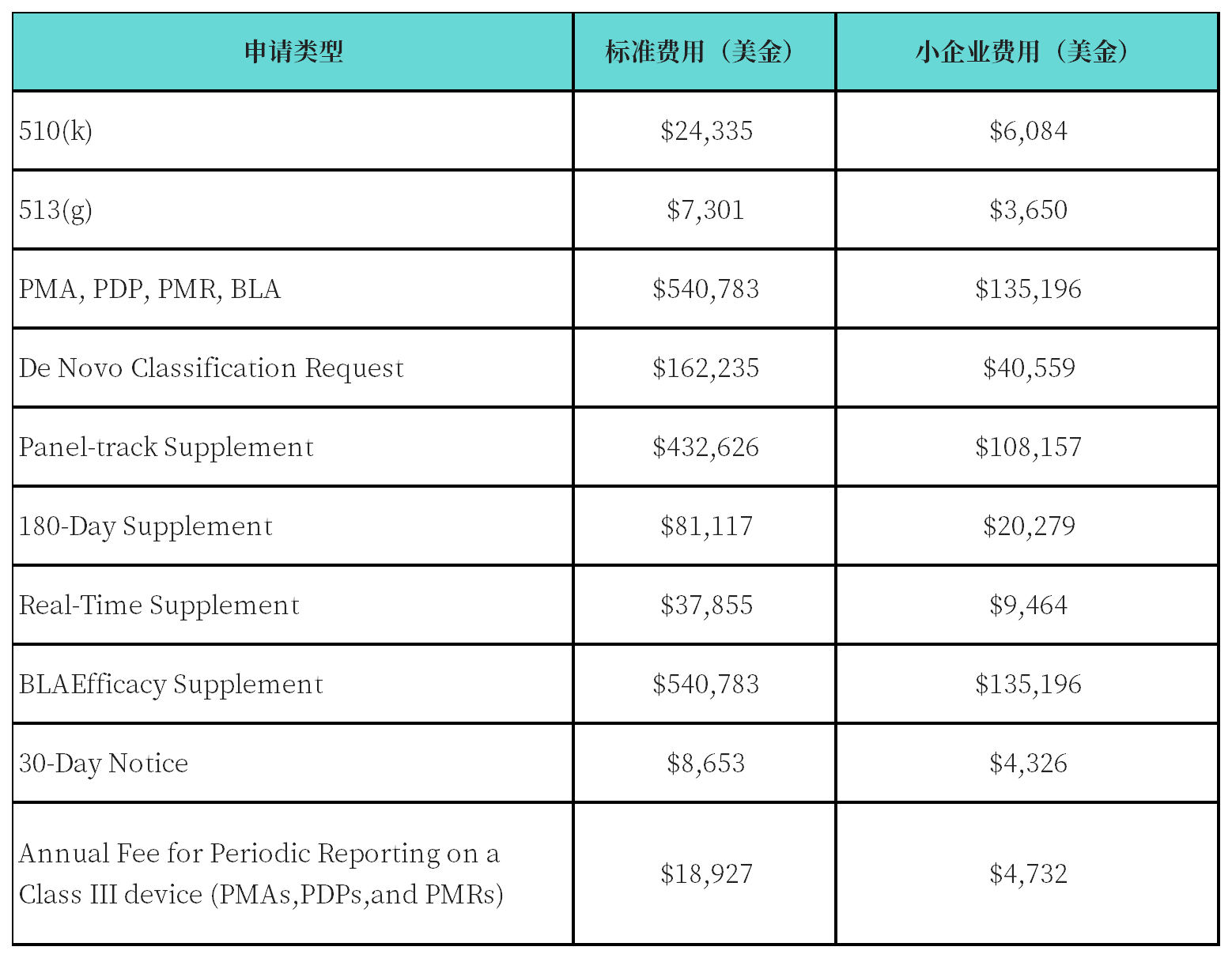
备注:以上费用所属年度为2024年10月1日至2025年9月30日