

联系我们
仲邈检测技术(上海)有限公司
电话:400-869-7268
孔老师:18101860670
李老师:18117149592
网址:www.shzmiao.com
邮箱:sales@shzmiao.cn
地址:上海市诸光路1588弄虹桥世界中心B栋一楼763号
欧盟CE认证
一、 欧盟监管
1. 监管部门

欧盟由27个成员国组成。参与医疗器械注册审评和监管活动的组织机构主要包括:公共卫生部、医疗器械协调小组、第三方验证机构(NB机构,Notify Body)与各国主管部门等。
NB机构(Notify Body)是欧盟按照新方法指令实施市场准入管理的重要技术实体,它们负责审核和验证产品是否符合欧盟指令的要求,确保产品安全和质量。
2. 相关法规
欧盟医疗器械法规(法规(EU)2017/745),自 2021 年 5 月 26 日起适用
体外诊断设备法规(法规(EU)2017/746),2022 年 5 月 26 日起适用。
2023 年3月 15 日欧洲议会和理事会 条例 (EU)2023/607 ,修订条例(EU)20171745 和 (EU)20171746,涉及某些医疗器械和体外诊断医疗器械的过渡规定。该法规对医疗器械(MDR)条例(EU)20171745 规定的过渡期进行了分阶段延长。
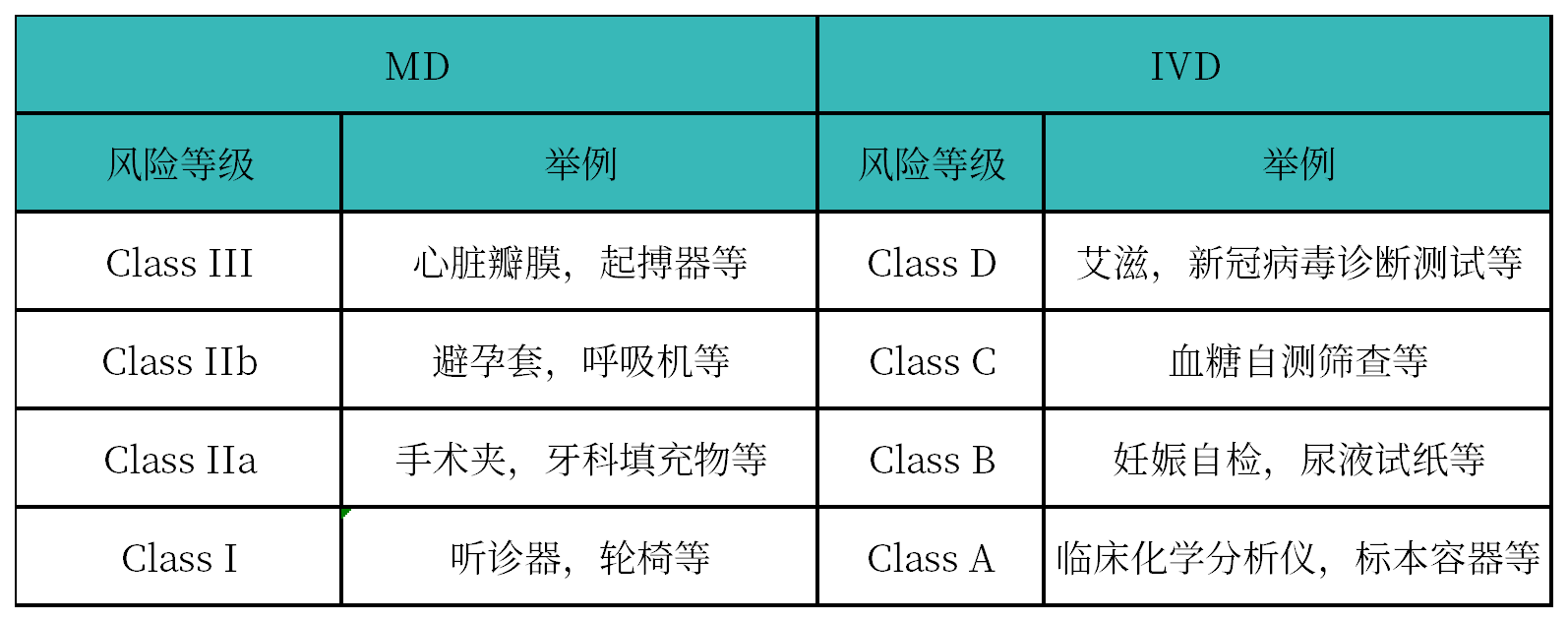
3. 风险等级分类
医疗器械分类:按风险级别从低到高、分为:I类(普通1类/灭菌Is/测量Im/重复使用Ir)、IIa、IIb和III类,其中I类(普通1类/灭菌Is/测量Im/重复使用Ir)为低风险,III类为高风险。
IVD分类:按风险级别从低到高分为 A、B、C、D类。
4. 准入必要条件
-
ISO 13485体系证书和年审报告
-
欧盟授权代表 European Authorized Representative
-
所有医疗器械都需要在EUDAMED数据库登记
5. 注册语言
英语
6. 欧盟授权代表职责
①市场准入:确保非欧盟制造商的产品符合欧盟的法规要求,包括技术文件准备、标签和说明书的合规性,协助非欧盟制造商获取市场准入,与欧洲各国的监管机构进行沟通和协商,以确保产品的合法销售和分销。
②通信与协调:作为制造商与欧洲监管机构之间的联系人,负责与监管机构就产品审批、注册、报告和其他问题进行沟通和协调,回答监管机构的查询,提供所需的技术文件,并确保制造商与监管机构之间的有效沟通。
③技术文件管理:负责在欧洲境内保存制造商的产品技术文件,并确保这些文件符合欧盟的法规要求,监督文件的准备、更新和维护,以便监管机构审查。
④产品监管:协助制造商遵守欧盟的产品监管要求,包括跟踪产品安全性和质量的相关事件,协助制造商进行市场监测和回收活动,确保及时报告和响应相关问题。
⑤验证合规性:验证制造商是否已起草欧盟符合性声明和是否进行了适当的合规性评估程序。
⑥文件保存:保存技术文件、合规性声明和相关证书的副本,并在监管机构要求时提供。
⑦注册义务:核实制造商已在EUDAMED数据库中注册了所需信息。
⑧合作与通知:与监管机构合作进行预防性和纠正性行动,并立即通知制造商有关投诉和监管机构对样品的请求。
⑨法律责任:如果制造商未遵守法规义务且不在欧盟境内,授权代表将与制造商共同承担因缺陷设备而产生的法律责任。
⑩终止授权:如果制造商违反其义务,授权代表应终止授权,并立即通知其所在地的成员国和相关认证机构。
这些职责确保了非欧盟制造商的医疗器械产品能够在欧盟市场上合法销售,同时符合欧盟的监管要求。
二、 注册流程|周期
1. 注册流程
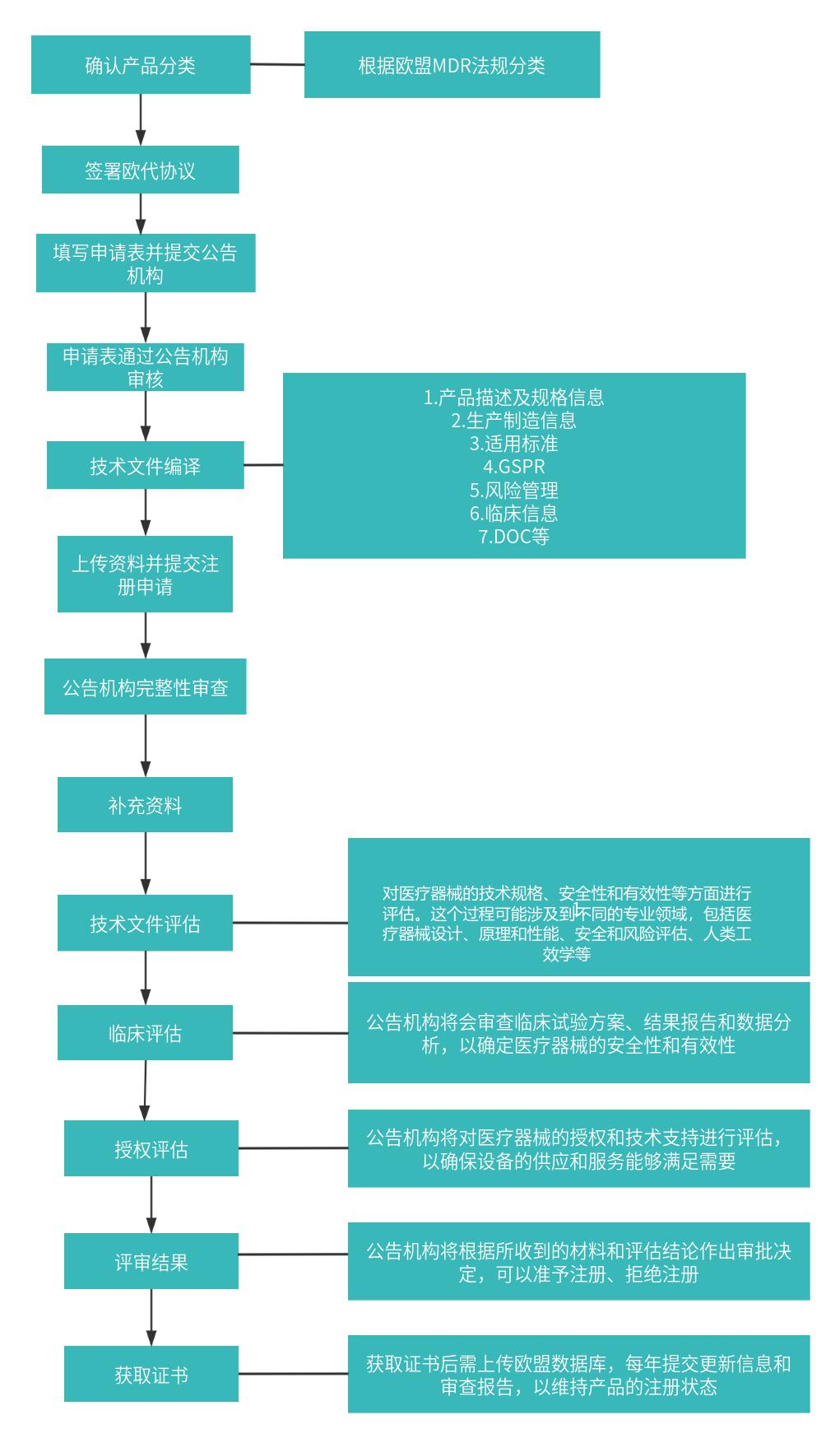
2. 注册周期
I类基本产品:根据我们的项目经验为1-2周,以实际为准。
Im,Ir、Is、Ia、IIb、 III 类产品:几个月至几年,一般情况为9-24个月,影响周期的因素如下:
①公告机构不同
②产品复杂程度不同
③产品风险等级不同
④产品注册文件基础材料不同